CFTR mutation leads to intrinsic dysfunction in neutrophils from people with Cystic Fibrosis
CFTR mutation leads to intrinsic dysfunction in neutrophils from people with Cystic Fibrosis
Robledo-Avila, F. H.; Rascon, R.; Montanez-Barragan, A.; Loyo-Celis, V.; Singh, H.; McCoy, K. S.; Kopp, B. T.; Partida-Sanchez, S.
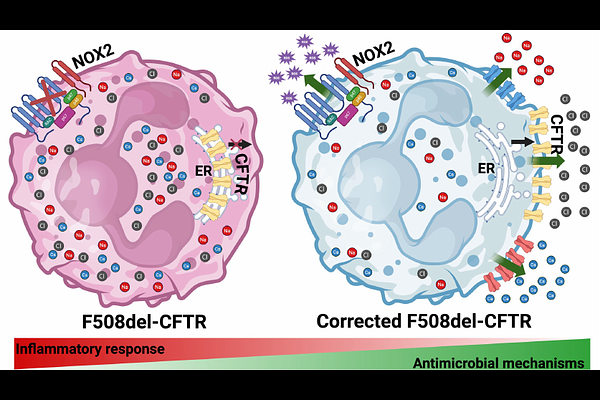
AbstractCystic fibrosis (CF), a common genetic disease, is caused by a defective CF-transmembrane conductance regulator (CFTR). People with CF (pwCF) are prone to develop infections by opportunistic pathogens, including Burkholderia cenocepacia, leading to chronic inflammation and lung function loss. Neutrophils, the most abundant cells in the chronically inflamed lungs of pwCF, release granular proteins and oxida-tive products that contribute to tissue damage. The CFTR modulators are a new treat-ment for pwCF aiming to correct the subcellular location and function of the CFTR ion channel. The triple modulator combination of Elexacaftor, Tezacaftor, and Ivacaftor (ETI) or Trikafta(R) has significantly improved clinical symptoms and overall provided a better quality of life for pwCF. The mechanism by which the CFTR modulators help to restore the antimicrobial functions of neutrophils is unknown. The present study demonstrates that neutrophils functionally express CFTR and reveals how ETI modi-fies subcellular CFTR trafficking in CF neutrophils. In addition, ETI treatment reduces intracellular chloride levels in human neutrophils, indicating activation of CFTR-dependent chloride efflux (outflow). Finally, ETI treatment also re-established the intra-cellular antimicrobial killing of CF neutrophils by potentiating NADPH oxidase activity and producing Neutrophil Extracellular Traps (NETs). Together, our findings suggest that CFTR has an essential role in controlling neutrophil functions and that the CFTR modulators improve the health of pwCF by restoring the antimicrobial functions of CF neutrophils.