Patient iPSC-Derived Cartilage Organoids Reveal Defective ECM Deposition and Altered Chondrogenic Trajectory in Saul-Wilson Syndrome
Patient iPSC-Derived Cartilage Organoids Reveal Defective ECM Deposition and Altered Chondrogenic Trajectory in Saul-Wilson Syndrome
Mahajan, S.; Ancel, S.; Ascone, G.; Kaur, R.; Torres, J.; Murad, R.; Wang, Y. X.; Ferreira, C. R.; Freeze, H.
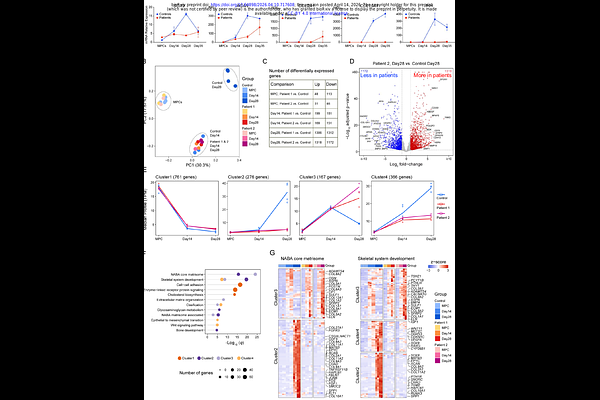
AbstractSaul-Wilson syndrome (SWS) is a skeletal dysplasia characterized by primordial dwarfism and progeroid features caused by a recurrent dominant COG4 variant (p.G516R). We previously showed that this mutation accelerates Golgi retrograde trafficking and disrupts glycosylation of the proteoglycan decorin, while zebrafish models revealed defects in chondrocyte elongation and intercalation. We have also shown that the SW1353 chondrosarcoma cells carrying the SWS variant exhibit reduced secretion of extracellular matrix (ECM) components. While these results indicate a critical function of COG4 in Golgi processing, the developmental process leading to skeletal dysplasia in SWS patients remains unknown. Here, we generated patient-derived iPSC cartilage organoids (SWS organoids), modeling early human chondrogenesis. SWS organoids failed to produce cartilage structures and displayed poor expression of chondrogenic markers. Time-course RNA-seq analysis of the chondrogenic process revealed reduced activation of gene networks involved in skeletal development, ECM organization, ossification, and glycosaminoglycan metabolism. Spatial multiomic analysis of protein and glycosylation by CODEX and GLYPH imaging revealed an altered chondrogenic trajectory, persistence of mesenchymal states, global glycosylation changes, and reduced deposition of chondroitin sulfate proteoglycans. These results indicate that the COG4 mutation disrupts ECM glycosylation and chondrogenic commitment, and that SWS organoids model early defects in cartilage formation underlies impaired skeletal growth in SWS.