Connective tissue growth in a mouse model of Kosaki overgrowth syndrome is limited by STAT1
Connective tissue growth in a mouse model of Kosaki overgrowth syndrome is limited by STAT1
Kim, J.; Kwon, H. R.; Berry, W.; Olson, L. E.
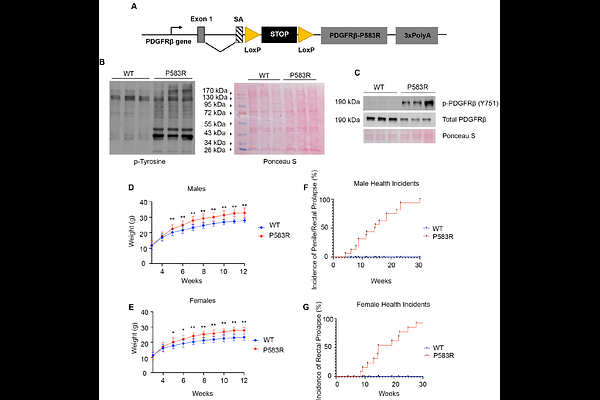
AbstractMutations in platelet-derived growth factor receptor beta (PDGFRb) cause Kosaki overgrowth syndrome (KOGS). Patients exhibit increased linear growth, craniosynostosis, and thin skin with increased elasticity and scarring. Of the KOGS patients identified to date, three unrelated individuals carried a P584R mutation in the juxtamembrane domain of PDGFRb, resulting in constitutive receptor activation. Due to the limited number of patients, extensive phenotyping and exploration of the molecular basis of disease, including modifier genes, has not been completed. We generated conditional knock-in mice to express mouse PDGFRb with a P583R mutation, corresponding to human P584R, under control of the endogenous Pdgfrb gene. Mutant mice were born at the expected ratio and appeared normal at birth. At 3 weeks of age, mutants began to exhibit connective tissue changes: increased body weight and bone length, craniosynostosis, ectopic bone in the tail and tendons, thin lipodystrophic skin, and high incidence of penile and rectal prolapse. To identify signaling changes caused by mutant PDGFRb signaling, we performed western blotting and phosphoproteomics on dermal fibroblasts. This uncovered increased phosphorylation of PDGFRb, PLCg, Akt1, Shp2, STAT1, STAT2, STAT3, and STAT5. Analysis of 6,621 proteins and 5,386 phosphopeptides identified upregulation of interferon signaling genes linked to STAT1. In many cell types, STAT1 has tumor-suppressor functions and acts to inhibit cell cycle. We generated Stat1-/- Pdgfrb+/P583R mice to test the contribution of STAT1 to KOGS phenotypes. Stat1-deletion exacerbated overgrowth and calvaria dysmorphogensis, and caused keloid-like skin fibrosis. No phenotypes present in the original Pdgfrb+/P583R mice were reverted to normal after Stat1 deletion. Therefore, the P583R mouse model mirrored KOGS phenotypes and increased activation of multiple PDGFRb signaling mediators; in this context, STAT1 activity opposes PDGFRb-driven overgrowth and fibrosis.