Rapid Assessment of Size, Shape, and Chemical Complementarity of Ligands for Computational Protein Design
Rapid Assessment of Size, Shape, and Chemical Complementarity of Ligands for Computational Protein Design
Petrenas, R.; Ozga, K.; Chubb, J. J.; Romanyuk, A. V.; McManus, J. J.; Leggett, G. J.; Scrutton, N. S.; Oliver, T. A. A.; Woolfson, D. N.
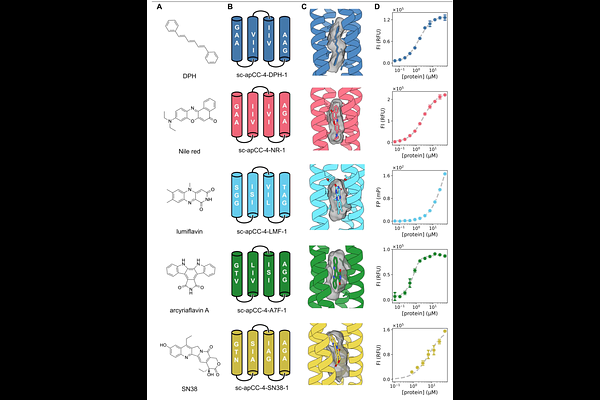
AbstractDriven by deep-learning approaches, computational protein design is advancing rapidly, and it is now possible to generate many de novo protein structures quickly and robustly. This sets new frontiers for the field, including designing proteins that bind small molecules tightly and specifically, and understanding the non-covalent interactions that underpin such designs to make binding predictable and tunable. Here we address these challenges with a rapid physics-based computational method to generate isosteric and chemically complementary binding pockets for small-molecule targets in de novo designed proteins. We test this experimentally by constructing and characterizing binding proteins for several synthetic and natural chromophores. By evaluating only single-digit numbers of designs, the pipeline delivers stable proteins with pre-organized binding sites confirmed by X-ray crystallography, which bind the targets selectively with micromolar affinities or better. To illustrate the scope and applications of this approach, we incorporate distinct and coupled chromophore-binding sites in a two-domain de novo protein enabling controlled energy transfer between the two sites, and we develop a small de novo binding protein that can be used in live mammalian cells to visualize sub-cellular structures.