Beyond Invariable Sites: Using Evolutionary Stasis to Map Multi-Layered Constraints on the Evolution of Viral and Mammalian Genomes
Beyond Invariable Sites: Using Evolutionary Stasis to Map Multi-Layered Constraints on the Evolution of Viral and Mammalian Genomes
Kosakovsky Pond, S. L.; Verdonk, H.; Weaver, S.; Brown, G.; Callan, D.; Nekrutenko, A.; Martin, D. P.
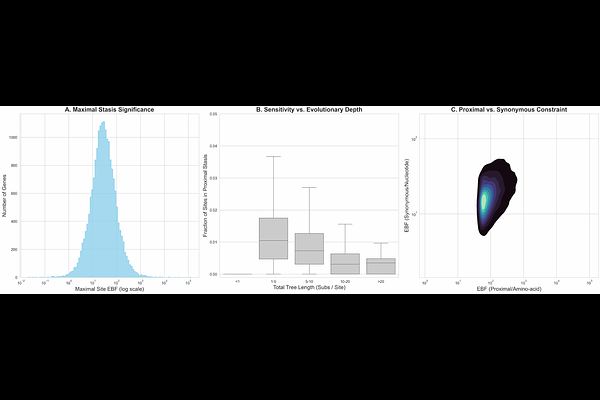
AbstractThe quantification of genomic conservation has progressed from foundational statistical modeling of evolutionary rates to state-of-the-art phylogeny-aware deep learning architectures. Yet, a fundamental resolution gap remains whenever evolutionary rates closely approach the "zero-rate origin," where standard selection inference tools will essentially ignore signals of extreme purifying section at invariant genome sites. We present B-STILL (Bayesian Significance Test of Invariant Low Likelihoods), a hierarchical Bayesian framework designed to resolve the selective landscape of protein-coding data by leveraging gene-level calibration and codon-site specific evolutionary opportunity. This framework is based on computationally efficient approximations using codon-substitution models which are scalable to alignments with thousands of sequences. By explicitly tuning the stasis radius around the near-zero evolutionary-rate regime, B-STILL distinguishes between stochastic invariance and functional constraint, identifying Evolutionary Stasis Anchors (ESAs) where the upper bound on permitted evolutionary change is statistically anomalous relative to the background of the gene. This hierarchical approach provides a signature of functional or structural constraint that is often difficult to detect using other tools. Validation against extensive pathogen and clinical databases confirms that ESAs are predictors of biological fitness and disease potential. Collectively, we identified thousands of significantly clustered ESAs that precisely footprint both known functional domains and currently uncharacterized structural motifs in mammalian and viral genomes. These findings establish B-STILL as a scalable statistical framework for high-resolution genomic annotation, ransforming formerly ignored invariant genome and protein sites into informative markers of extreme purifying selection across both well-characterized and uncharacterized protein-coding genes from different domains of life.