CGRig: a rigid-body protein model with residue-level interaction sites for long-time and large-scale protein assembly simulation
CGRig: a rigid-body protein model with residue-level interaction sites for long-time and large-scale protein assembly simulation
Teshirogi, Y.; Terada, T.
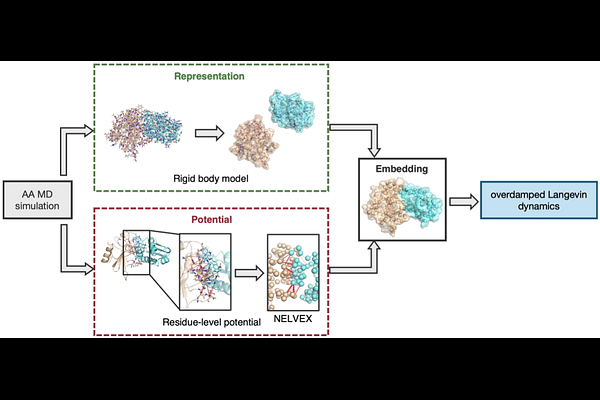
AbstractMolecular dynamics (MD) simulations are a powerful tool for investigating biomolecular dynamics underlying biological functions. However, the accessible spatiotemporal scales of conventional all-atom simulations remain limited by high computational costs. Coarse-graining reduces these costs by decreasing the number of interaction sites and enabling longer timesteps. In extreme cases, proteins are represented as single spherical particles; while such approximations facilitate cellular-scale simulations, they often sacrifice essential structural information, such as molecular shape and interaction anisotropy. Here, we present CGRig, a rigid-body protein model with residue-level interaction sites designed for long-time, large-scale simulations. In CGRig, each protein is treated as a single rigid-body embedding residue-level interaction sites. Its translational and rotational motions are described by the overdamped Langevin equation incorporating a shape-dependent friction matrix. Intermolecular interactions are calculated using G[o]-like native contact potentials, Debye-Huckel electrostatics, and volume exclusion. We validated that CGRig accurately reproduces the translational and rotational diffusion coefficients expected from the friction matrix for an isolated protein. For dimeric systems, the model successfully maintained native complex structures. Furthermore, two initially separated proteins converged into the correct complex with an association rate consistent with all-atom simulations. Notably, CGRig achieved a simulation performance exceeding 17 s/day for a 1,024-molecule system. These results demonstrate that CGRig provides an efficient framework for simulating protein assembly while retaining residue-level interaction specificity, making it a valuable tool for investigating large-scale biomolecular self-assembly.