Interaction between long-range chromatin regulators Nipbl & Isl1 synergistically drives heart defects in mice
Interaction between long-range chromatin regulators Nipbl & Isl1 synergistically drives heart defects in mice
Chea, S.; Santos, R.; Lopez-Burks, M. E.; Lander, A.; Calof, A.
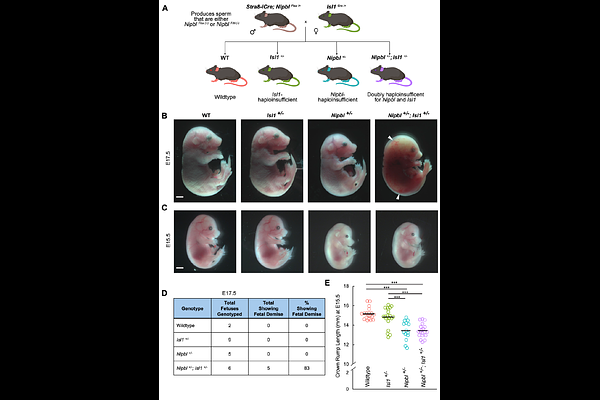
AbstractCongenital heart defects (CHDs) are frequently observed in the most common form of Cornelia de Lange Syndrome (CdLS), which is caused by haploinsufficiency for NIPBL, a gene involved in chromatin looping and cis-regulatory control of gene expression. Here, we surveyed cardiac defects in mice made Nipbl-haploinsufficient in the second heart field using two Cre drivers: Mef2c-Cre and Isl1-Cre. Only Isl1-Cre-driven Nipbl-haploinsufficiency resulted in CHDs - a finding we traced to the additional contribution of Isl1-haploinsufficiency caused by the Isl1-Cre allele. To test whether combined reduction of Nipbl and Isl1 cause CHDs, we made mice globally haploinsufficient for both genes. Indeed, Nipbl+/-; Isl1+/- mice exhibited a substantially higher frequency and severity of CHDs than mice haploinsufficient for either gene alone. As a member of the LIM-homeodomain transcription factor family, Isl1 is involved in chromatin looping and enhancer-promoter communication via a mechanism distinct from that of Nipbl. Nevertheless, when we performed RNA sequencing on E10.5 hearts from wildtype, Nipbl+/-, Isl1+/-, and Nipbl+/-; Isl1+/- embryos, we observed that combined haploinsufficiency resulted in largely additive gene expression changes, including dysregulation of known cardiac regulators (Irx4, Tbx1, Foxo6, Heyl, Bnc1, Sox17) and novel candidates (Gbx1, Csdc2, Myrf, Pou6f1, Zfp579, ad Zfp763). A subset of additive changes arose from opposing regulatory influences in single mutants that restored gene expression to WT levels in Nipbl/-; Isl1/- hearts. For example, Hoxc4, Pitx2, Isl1 itself, and Pax6 (a known target of Isl1), were upregulated in Nipbl+/- hearts, downregulated in Isl1+/- hearts, but expressed at WT levels in Nipbl/-; Isl1/- hearts. Since loss of Isl1 upregulation from Nipbl+/- to Nipbl+/-; Isl1+/- hearts coincided with a marked increase in CHDs, we propose that Isl1 upregulation compensates for the loss of cis-regulatory interactions due to Nipbl-haploinsufficiency, and protects hearts from severe CHD risk. Supporting this model, other LIM-homeodomain transcription factors (Lhx2, Lhx3, Lhx9) were also upregulated in Nipbl+/- hearts, with Lhx3 and Lhx9 showing even greater upregulation in Nipbl+/-; Isl1+/- hearts. Despite this, CHDs resulting from the combined loss of Nipbl and Isl1 were particularly severe. These findings suggest that heart development is exquisitely sensitive to small change in gene expression, leading to synergistic phenotypic interactions when relatively modest gene expression changes are combined.