TDP-43 toxic gain of function links ALS/FTLD-TDP and Alzheimer's Disease through splicing
TDP-43 toxic gain of function links ALS/FTLD-TDP and Alzheimer's Disease through splicing
Van Zuiden, W.; Meimoun, T. D.; Bar, C.; Siany, A.; Moshe, L.; Yacovzada, N. S.; Weizman, E.; Neumann, M.; Buchman, A. S.; Wang, Y.; Bennett, D. A.; Glass, J. D. G. D.; Trautwig, A. N.; Seyfried, N.; Cooper-Knock, J.; Hornstein, E.
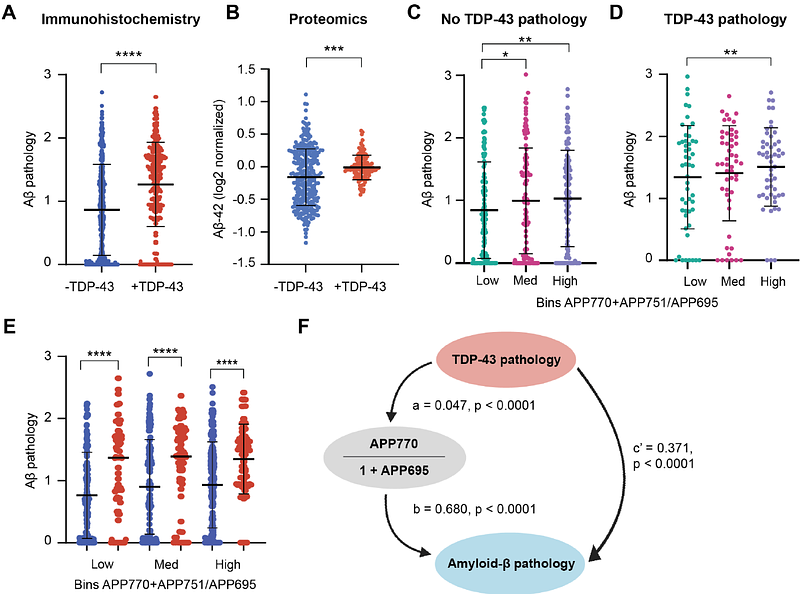
AbstractLoss of nuclear TDP-43 splicing activity is a common feature across neurodegenerative diseases, but its relevance to Alzheimer\'s disease (AD) remains unclear. Here, we show that TDP-43 pathology in AD is broadly associated with splicing abnormalities, including aberrant splicing of amyloid precursor protein (APP). We demonstrate that TDP-43 drives the formation of elongated APP isoforms, APP751 and APP770. Thus, TDP-43 dysregulation disrupts APP695/751/770 alternative splicing across ALS/FTLD-TDP and AD, providing a compelling mechanism for a 37-year-old observation of APP isoform dysregulation. We further establish a mechanistic link between TDP-43 pathology, APP splicing, and A{beta} pathology. Unexpectedly, this effect is mediated by a toxic gain of cytoplasmic TDP-43 function, rather than loss of its nuclear role. Using proximity proteomics and base editing in human iPSC-derived neurons, we show that TDP-43 pathology causes cytoplasmic co-sequestration of splicing regulators SCAF11, SRSF5, and TIAL1, which are involved in APP mis-splicing, but not in the regulation of other TDP-43 targets such as STMN2 or UNC13A. Together, our findings suggest that TDP-43-mediated splicing dysfunction upstream of APP contributes to the pathogenesis of seemingly disparate neurodegenerative diseases, uniting AD and ALS/FTLD-TDP through a shared molecular mechanism.