It's a wrap: deriving distinct discoveries with FDR control after a GWAS pipeline
It's a wrap: deriving distinct discoveries with FDR control after a GWAS pipeline
Chu, B.; He, Z.; Sabatti, C.
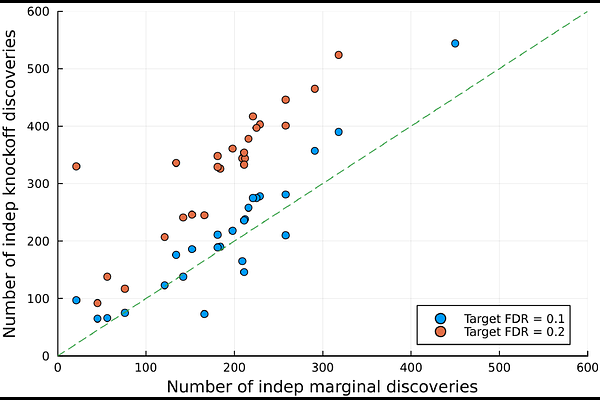
AbstractRecent work has shown how to test conditional independence hypotheses between an outcome of interest and a large number of explanatory variables with false discovery rate control (FDR), even without access to individual level data. In the case of genome-wide association studies (GWAS) specifically, summary statistics resulting from the standard analysis pipeline can be used as input of a procedure which identifies distinct signals across the genome with FDR control. This secondary analysis requires sampling of negative controls (knockoff) from a distribution determined by the linkage disequilibrium patterns in the genome of the population under study. In prior work, we have pre-computed this distribution for European genomes, starting from information derived from the UK Biobank. Thus, researchers working with European GWASes can carry out a knockoff analysis with minimal computational costs, using the distributed routine GhostKnockoffGWAS. Here we introduce and release a new software (solveblock) that extends this capability to a much richer collection of studies. Given a set of genotyped samples, or a reference dataset, our pipeline efficiently estimates the high-dimensional correlation matrices that describe correlation structures across the genome, making rather common sparsity assumptions. Taking this sample-specific estimate as input, the software identifies groups of genetic variants that are highly correlated, and uses them to define an appropriate resolution for conditional independence hypotheses. Finally, we compute the distribution for the exchangeable negative controls necessary to test these hypotheses. The output of solveblock can be passed directly to GhostKnockoffGWAS, allowing users to carry out the complete analysis in a two step procedure. We illustrate the performance of the routine analyzing data from five UK Biobank sub-populations. In simulations, our method controls FDR. Analyzing real data relative to 26 phenotypes of varying polygenicity in British individuals, we make an average of ~19 additional discoveries, compared to standard marginal association testing. Our code, precompiled software, and processed files for these five sub-populations are openly shared.