Snekmer Learn/Apply: A kmer-based vector similarity approach to proteinclassification suitable for metagenomic datasets
Snekmer Learn/Apply: A kmer-based vector similarity approach to proteinclassification suitable for metagenomic datasets
Nitka, T. A.; Jacobson, J.; Chang, C. H.; Krause, G. R.; Wheeler, T. J.; Egbert, R. G.; Nelson, W. C.; McDermott, J. E.
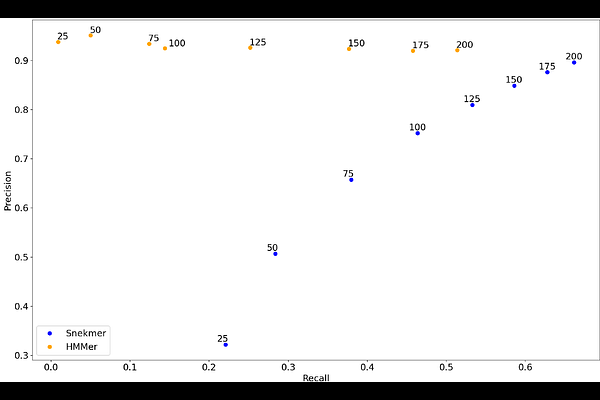
AbstractAdvances in whole genome sequencing have led to a rapid and ongoing increase in the amount of sequence data available, but 40-50% of known genes have no functional annotation and only 25-30% have specific functional annotations. Current functional annotation approaches typically rely on computationally expensive pairwise or multiple sequence alignments, preventing rapid development of models for novel protein functions and sometimes limiting methods to one ontology. Representation of sequence in short segments (kmers) has been used in many applications for nucleotide sequence, and more recently has been applied to protein sequence as well. We previously developed Snekmer, a tool which uses kmer patterns to develop alignment-free individual protein family models. Other approaches, such as MMSeqs2 and DIAMOND, use protein kmers as a fast filter to reduce search space for subsequent sequence alignment. Here, we describe a novel addition to the Snekmer tool which builds kmer libraries for protein families and uses those libraries to map functional annotations to new sequences. We first demonstrate that our method accurately applies TIGRFAMs annotations to protein fragments and to a low-sequence identity benchmark dataset, and further use it to annotate a set of drought stress associated soil and rhizosphere metagenome sequences with higher sensitivity towards several important protein function classes than that shown by HMMs. We have incorporated this workflow into Snekmer.