Identification of reactive CpGs and RNA expression in early COVID-19 through cis-eQTM analysis reflecting disease severity and recovery
Identification of reactive CpGs and RNA expression in early COVID-19 through cis-eQTM analysis reflecting disease severity and recovery
Ryu, H.; An, K.; Kwon, Y.; Jeon, Y.; Jeon, S.; Choi, H.; Kim, Y. J.; Kim, S.; Sul, O. J.; Lee, S.; Chun, A. Y.; Shin, E.-S.; Ra, S. W.; Bhak, J.
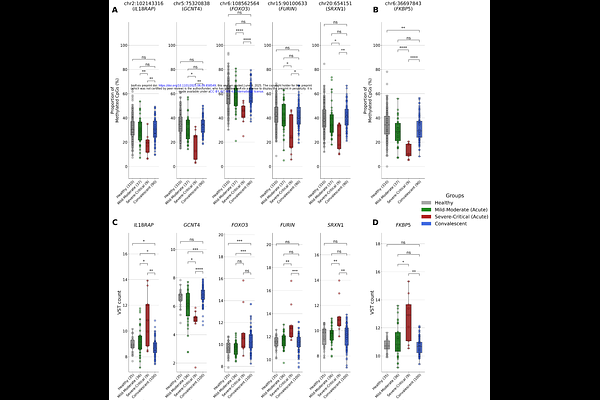
AbstractMulti-omics analyses of severe COVID-19 cases are crucial in deciphering the complex interplay between genetic and epigenetic factors. Here, we present an analysis of Expression Quantitative Trait Methylation (eQTM) to investigate the complex interplay of methylation and gene expression pattern during the acute phase of severe COVID-19. We identified 16 differentially expressed genes and 30 nearby differentially methylated CpG sites. Six key genes SRXN1, FURIN, IL18RAP, FOXO3, GCNT4, and FKBP5 were either up-regulated or down-regulated near hypomethylated CpG sites. These genes are associated with viral infiltration, immune activation, lung damage, and oxidative stress-related multi-organ failure, which are the hallmarks of severe COVID-19. Interestingly, during the recovery phase, methylation and gene expression levels returned to baseline, underscoring the rapid and reversible nature of these molecular changes. These findings provide insight into the dynamics of epigenetic and transcriptomic shifts according to the infectious stage, supporting potential prognostic and therapeutic approaches for severe COVID-19.