Simpatico: accurate and ultra-fast virtual drug screening with atomic embeddings
Simpatico: accurate and ultra-fast virtual drug screening with atomic embeddings
Gaiser, J.; Wheeler, T. J.
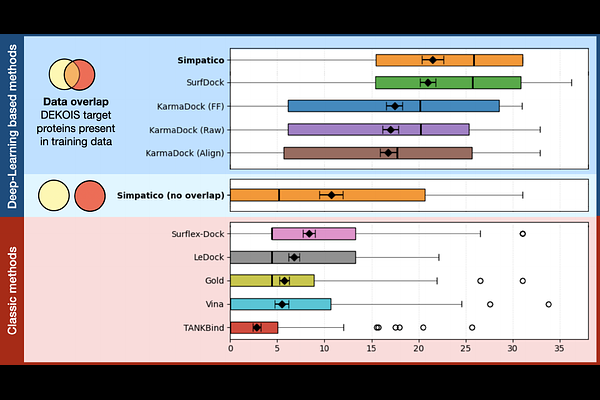
AbstractBuilding on established methods for molecular docking, structure-based deep learning has recently yielded important advances in virtual drug screening. We present simpatico, a method that follows an alternate approach, based on the field of Representation Learning, to dramatically speed the process of accurate drug screening. Simpatico employs graph neural networks to produce high-dimensional embeddings for the atoms of proteins and small molecules, and uses these embeddings to rapidly produce accurate predictions of the interaction potential for drug candidates with target protein pockets. Simpatico can search a database containing 600 million drugs for good binding candidates to a single protein pocket in 2.5 hours on a single GPU. Despite being >1000x faster than state of the art docking and diffusion-based methods, simpatico is competitive with the most accurate of those methods. We also observe that simpatico embeddings can be used to explore toxicity risk and to identify proteins with similar binding potential. Simpatico is open source software; all code, weights, and data may be accessed at https://github.com/TravisWheelerLab/Simpatico.