SETD2 suppresses tumorigenesis in a KRASG12C-driven lung cancer model and its catalytic activity is regulated by histone acetylation
SETD2 suppresses tumorigenesis in a KRASG12C-driven lung cancer model and its catalytic activity is regulated by histone acetylation
Mack, R.; Flores, N. M.; Fox, G. C.; Dong, H.; Cebeci, M.; Hausmann, S.; Chasan, T.; Dowen, J.; Strahl, B. D.; Mazur, P. K.; Gozani, O.
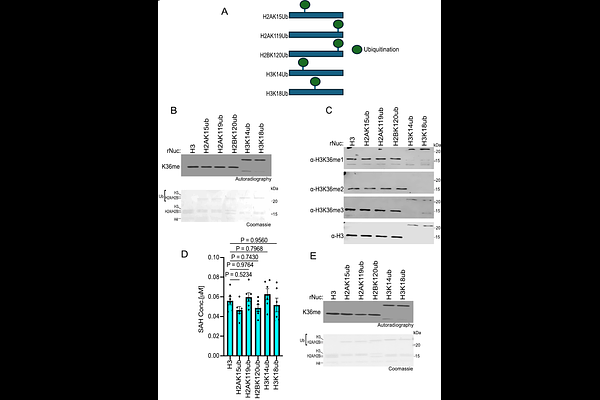
AbstractHistone H3 trimethylation at lysine 36 (H3K36me3) is a key chromatin modification that regulates fundamental physiologic and pathologic processes. In humans, SETD2 is the only known enzyme that catalyzes H3K36me3 in somatic cells and is implicated in tumor suppression across multiple cancer types. While there is considerable crosstalk between the SETD2-H3K36me3 axis and other epigenetic modifications, much remains to be understood. Here, we show that SETD2 functions as a potent tumor suppressor in a KRASG12C-driven lung adenocarcinoma (LUAD) mouse model, and that acetylation at H3K27 (H3K27ac) enhances SETD2 in vitro methylation of H3K36 on nucleosome substrates. In vivo, SETD2 ablation accelerates lethality in an autochthonous KRASG12C-driven LUAD mouse tumor model. Biochemical analyses reveal that polyacetylation of histone tails in a nucleosome context promote H3K36 methylation by SETD2. In addition, monoacetylation exerts position-specific effects, such as H3K27ac stimulating SETD2 methylation activity. In contrast, mono-ubiquitination at various histone sites, including at H2AK119 and H2BK120, does not affect SETD2 methylation of nucleosomes. Together, these findings provide insight into how SETD2 integrates histone modification signals to regulate H3K36 methylation and highlights the potential role of SETD2-associated epigenetic crosstalk in cancer pathogenesis.