Multimodal deep learning integration of cryo-EM and AlphaFold3 for high-accuracy protein structure determination
Multimodal deep learning integration of cryo-EM and AlphaFold3 for high-accuracy protein structure determination
Gyawali, R.; Dhakal, A.; Cheng, J.
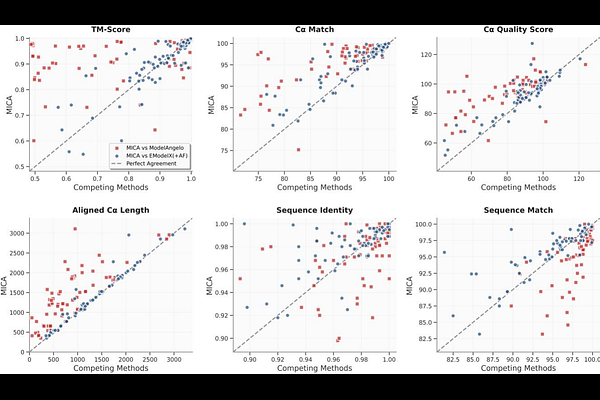
AbstractCryo-electron microscopy (cryo-EM) is a key technology for determining the structures of proteins, particularly large protein complexes. However, automatically building high-accuracy protein structures from cryo-EM density maps remains a crucial challenge. In this work, we introduce MICA, a fully automatic and multimodal deep learning approach combining cryo-EM density maps with AlphaFold3-predicted structures at both input and output levels to improve cryo-EM protein structure modeling. It first uses a multi-task encoder-decoder architecture with a feature pyramid network to predict backbone atoms, C atoms and amino acid types from both cryo-EM maps and AlphaFold3-predicted structures, which are used to build an initial backbone model. This model is further refined using AlphaFold3-predicted structures and density maps to build final atomic structures. MICA significantly outperforms other state-of-the-art deep learning methods in terms of both modeling accuracy and completeness and is robust to protein size and map resolution. Additionally, it builds high-accuracy structural models with an average template-based modeling score (TM-score) of 0.93 from recently released high-resolution cryo-EM density maps, showing it can be used for real-world, automated, accurate protein structure determination.