A residual-ratio framework for auditing transcriptomic gene signatures against background expression structure
A residual-ratio framework for auditing transcriptomic gene signatures against background expression structure
Zhu, Y.; Zhang, C.; Calhoun, V. D.; Bi, Y.
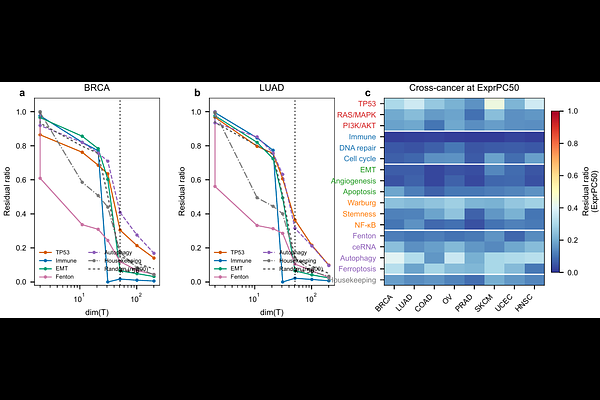
AbstractBackground: Transcriptomic gene signatures are widely used to infer pathway activity and biological mechanism from bulk cancer expression data, yet current evaluation strategies primarily emphasize internal coherence, predictive performance, or scoring robustness. A quantitative framework for assessing how much signature variation remains independent of background expression structure has been lacking. Results: Unlike existing single-number signature-quality metrics such as Berglund uniqueness, residual-ratio auditing reports a trajectory across null-model richness: for each signature we compute the residual ratio resratio(k) = 1 - sum_{j=1}^{k}(mathbf{q}_j^top mathbf{h})^2 at progressively enriched expression-PC subspaces, together with an inverse-participation-ratio (IPR) concentration diagnostic that reports the effective number of axes absorbing each signature. Applied to a curated 17-entry benchmark, all 50 MSigDB Hallmark gene sets, and 1181 Reactome pathways across 8 TCGA cancer types (4462 samples), with external validation in METABRIC, the framework produces two complementary readouts. First, the curated panel is absorbed into the ExprPC50 subspace at residual ratios 18--43% below size-matched random 30-gene baselines in every cancer (curated mean resratio range 0.109--0.177 vs. random mean 0.18--0.288), providing the framework's central quantitative discrimination between biologically coherent signatures and arbitrary gene combinations. Second, within the curated panel the ExprPC50 residual ratio is negatively correlated with the top-5 absorption concentration in every cancer (Spearman rho from -0.59 in PRAD to -0.89 in SKCM, median -0.71; all 8 significant at p < 0.05, most at p < 10^-3); we report this correlation as a descriptive geometric property of the null-model coordinate system rather than as a biological law, because 1000 random 30-gene draws projected through the same top-50 expression-PC basis reproduce the same pan-cancer median rho (-0.73; Supplementary Table~ref{tab:S17}), and it is robust to compositional nuisance: after rebuilding the null basis as immune-PC1 oplus stromal-PC1 oplus proliferation-PC1 plus 47 residual PCs, the per-cancer rho becomes more negative rather than shallower (median -0.86; Supplementary Table~ref{tab:S18}), ruling out tumor purity, immune infiltrate, and stromal fraction as drivers of the pattern. Because absorption at ExprPC50 is a geometric property of how any signature direction sits in expression-PC space, tier-level distributional structure at this operating point is not separable beyond the low-vs-upper band split: a Kruskal--Wallis omnibus is significant (p = 4.9 x 10^-13), but pairwise Dunn's post-hoc tests show that Tiers~1, 4, and~5 are not separable (p_{\mathrm{BH}} > 0.2). The trajectory shape itself is empirically bootstrap-invariant: across 200 sample-level fixed-basis bootstrap resamples of the 17 curated entries in BRCA, the mean pairwise Pearson correlation of trajectory-shape vectors is 0.999, and individual cell-level 95% bootstrap CI half-widths at B = 1000 resamples are in the range 0.002--0.053. External replication in the METABRIC breast cancer cohort (n_{\text{samples}} = 1980, microarray) showed moderate-to-strong rank-ordering concordance with TCGA-BRCA across the 17 curated entries (Spearman rho = 0.72 on the 17-signature ordering, 95% Fisher-z CI 0.37--0.89, p = 0.001). Under an upper-bound sensitivity analysis, 45 of 50 Hallmark gene sets and 992 of 1181 Reactome pathways had ExprPC200 residual ratios below the mean of their size-matched random baselines---a descriptive statistic reflecting axis alignment under rich null models, not a failure rate. In causal DAG simulations (n_{mathrm{rep}} = 100 replicates), a signature driven entirely by a latent confounder retained resratio = 0.233 at ExprPC50, numerically comparable to Tier~1 validated drivers, so a single-point residual ratio cannot adjudicate confounder-independence. The framework's load-bearing signals are therefore the trajectory shape (statistically invariant under sample-level resampling) and the magnitude gap between the curated panel and its random 30-gene baseline (the curated-vs-random discrimination), read jointly---not the value of resratio at any single null-model dimensionality. Conclusions: Residual-ratio auditing provides an interpretable and practical framework for quantifying how much of a transcriptomic gene signature's variance remains orthogonal to a chosen background-expression model. The two statistically reliable quantities it reports are (i) the shape of the trajectory resratio(k) across null-model richness, which is bootstrap-invariant across sample-level resamples, and (ii) the magnitude gap between the curated panel's residual ratio and size-matched random 30-gene baselines at a fixed operating point, which is 18-43% in all 8 TCGA cancers and survives a purity-aware null-model construction. The negative correlation between resratio and the top-5 absorption concentration $c$ (curated-panel median rho = -0.71) is reproduced by random 30-gene sets under the same basis (random-draw median rho = -0.73) and is therefore best read as a descriptive geometric property of the null-model coordinate system rather than a biological discovery about curated signatures. Any single operating-point residual ratio carries materially wider cell-level uncertainty than the trajectory shape and cannot, on its own, adjudicate confounder-independence. The framework's outputs describe a signature's geometric relationship to modeled background expression structure and do not evaluate clinical utility: a signature with a low residual ratio may still be clinically valuable when that low value reflects alignment with a strong prognostic or actionable program such as proliferation, immune infiltration, or cell cycle, and the framework is not a substitute for calibrated prognostic or predictive classifiers. All findings are based on bulk RNA-seq (TCGA PanCancer Atlas, 8~cancer types) and microarray (METABRIC) data; transfer to single-cell, single-nucleus, or spatial transcriptomics is out of scope and not claimed. Used within this scope---reading the trajectory shape and the magnitude-gap signal jointly, rather than the value of resratio at any one k---the framework adds a complementary audit layer to existing pathway-scoring and experimental-validation workflows, and supports more calibrated interpretation, comparison, and reporting of transcriptomic gene signatures in cancer studies.