The limits of Bayesian estimates of divergence times in measurably evolving populations
The limits of Bayesian estimates of divergence times in measurably evolving populations
Ivanov, S.; Fosse, S.; dos reis, M.; Duchene, S.
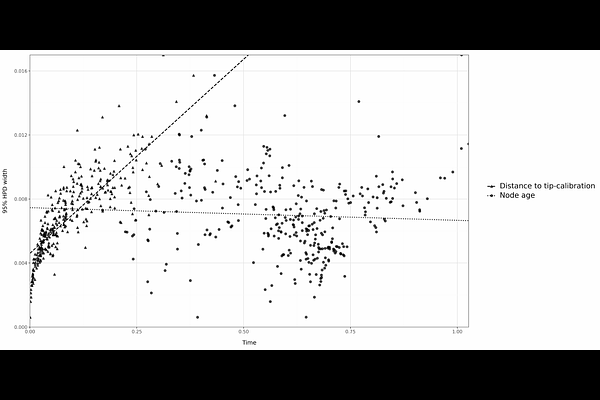
AbstractBayesian inference of divergence times for extant species using molecular data is an unconventional statistical problem: Divergence times and molecular rates are confounded, and only their product, the molecular branch length, is statistically identifiable. This means we must use priors on times and rates to break the identifiability problem. As a consequence, there is a lower bound in the uncertainty that can be attained under infinite data for estimates of evolutionary timescales using the molecular clock. With infinite data (i.e., an infinite number of sites and loci in the alignment) uncertainty in ages of nodes in phylogenies increases proportionally with their mean age, such that older nodes have higher uncertainty than younger nodes. On the other hand, if extinct taxa are present in the phylogeny, and if their sampling times are known (i.e., `heterochronous' data), then times and rates are identifiable and uncertainties of inferred times and rates go to zero with infinite data. However, in real heterochronous datasets (such as viruses and bacteria), alignments tend to be small and how much uncertainty is present and how it can be reduced as a function of data size are questions that have not been explored. This is clearly important for our understanding of the tempo and mode of microbial evolution using the molecular clock. Here we conducted extensive simulation experiments and analyses of empirical data to develop the infinite-sites theory for heterochronous data. Contrary to expectations, we find that uncertainty in ages of internal nodes scales positively with the distance to their closest tip with known age (i.e., calibration age), not their absolute age. Our results also demonstrate that estimation uncertainty decreases with calibration age more slowly in data sets with more, rather than fewer site patterns, although overall uncertainty is lower in the former. Our statistical framework establishes the minimum uncertainty that can be attained with perfect calibrations and sequence data that are effectively infinitely informative. Finally, we discuss the implications for viral sequence data sets. In a vast majority of cases viral data from outbreaks is not sufficiently informative to display infinite-sites behaviour and thus all estimates of evolutionary timescales will be associated with a degree of uncertainty that will depend on the size of the data set, its information content, and the complexity of the model. We anticipate that our framework is useful to determine such theoretical limits in empirical analyses of microbial outbreaks.