difFUBAR: Scalable Bayesian comparison of adaptive evolution
difFUBAR: Scalable Bayesian comparison of adaptive evolution
Sadiq, H.; Truong, P.; Danielsson, M.; Kumar, V.; Nordlinder, H. N.; Martin, D. P.; Murrell, B.
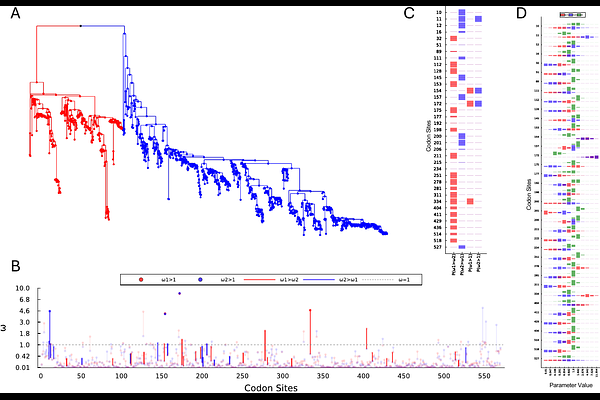
AbstractWhile many phylogenetic methods exist to characterize evolutionary pressure at individual codon sites, relatively few allow direct comparison between different a priori selected sets of branches. Such comparisons may be useful for pinpointing precisely the codon sites that are under differing selective pressures due to differing environmental contexts, or differing genomic contexts via epistatic interactions. Indeed, this was only recently addressed by an approach, developed in the frequentist framework, that proposes a site-wise likelihood ratio hypothesis test. Previously, we have demonstrated that approximate grid-based Bayesian approaches to characterizing site-wise variation in selection parameters can outperform individual site-wise likelihood ratio tests. Such grid-based approaches can exhibit poor computational scaling when the number of site-wise parameters expands, but here we show that this is still tractable up to four parameters, and that a simple subtree-likelihood caching strategy can provide efficiency improvements in some cases. We propose difFUBAR, which allows the demarcation of two branch sets of interest and, optionally, a background set, and estimates joint site-specific posterior distributions over , {omega}1, {omega}2, and {omega}BG using a Gibbs sampler. Evidence for hypotheses of interest can then be quantified directly from the posterior distribution, and we standardly report P({omega}1>{omega}2), P({omega}2>{omega}1), P({omega}1>1), and P({omega}2>1). We characterize the computational and statistical performance of this approach on previous simulations, comparing it to a site-wise likelihood ratio test approach, where it shows moderate statistical benefits, and substantial computational gains, typically being more than two orders of magnitude faster on the same datasets. We also demonstrate that it can scale to datasets of over ten thousand taxa, on a laptop in under ten minutes. difFUBAR is implemented in MolecularEvolution.jl - a Julia framework for phylogenetic model development - and can be run locally, or online via a Colab notebook.