AbDesign: Database of point mutants of antibodies with associated structures reveals poor generalization of binding predictions from machine learning models.
AbDesign: Database of point mutants of antibodies with associated structures reveals poor generalization of binding predictions from machine learning models.
Janusz, B.; Chomicz, D.; Demharter, S.; Arts, M.; Kanter, J. d.; Wilke, Y.; Britze, H.; Wrobel, S.; Gawlowski, T.; Dudzic, P.; Ukkivi, K.; Peil, L.; Spreafico, R.; Krawczyk, K.
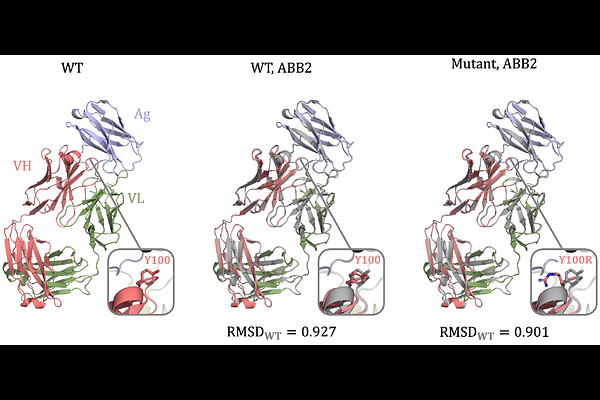
AbstractAntibodies are naturally evolved molecular recognition scaffolds that can bind a variety of surfaces. Their designability is crucial to the development of biologics, with computational methods holding promise in accelerating the delivery of medicines to the clinic. Modeling antibody-antigen recognition is prohibitively difficult, with data paucity being one of the biggest hurdles. Current affinity datasets comprise a small number of experimental measurements, which are often not standardized between molecules. Here, we address these issues by creating a dataset of seven antigens with two antibodies each, for which we introduce a heterogeneous set of mutations to the CDR-H3 measured by ELISA. Each of the parental complexes has a known crystal structure. We perform benchmarking of state-of-the-art affinity prediction algorithms to gauge their effectiveness. Current computational methods exhibit significant limitations in accurately predicting the effects of single-point mutations. In contrast, the older empirical, physics-based method FoldX, performs well in identifying mutants that retain binding. These findings highlight the need for more resources like the one presented here - large, molecularly diverse, and experimentally consistent datasets.